Figure 3.1--Structure of Pyridostigmine Bromide
Figure 3.1--Structure of Pyridostigmine Bromide
Much of this chapter is technical. Information in this chapter is relevant to understanding some aspects of why PB was given and what was known about its safety and efficacy prior to the PGW. The rationale for treatment with PB is discussed briefly but does not constitute a focus of this report. Other documents focus more specifically on the rationale for use (Pyridostigmine, 1996; Pope, 1997) and the limitations in this rationale (Prendergast, 1997).
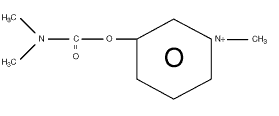
PB is a positively charged quaternary ammonium carbamate compound, which binds to ("carbamylates") two sites on acetylcholinesterase (AChE), a negatively charged site and an esteratic site, preventing acetylcholine (ACh) from binding to these same sites and being broken down by the enzyme (see Figure 3.1). This inhibition leads to buildup of ACh, which is not being hydrolyzed, resulting in increased action of ACh on its receptor.
Acetylcholine Receptors. Not all ACh receptors are alike. Receptors belong to one of two groups, "nicotinic" and "muscarinic," so termed because of specific chemicals--nicotine and muscarine--initially shown to bind to these receptors. Both types of receptors occur both in the CNS (central nervous system, including the brain and spinal cord) and peripherally (as part of the "PNS," or peripheral nervous system, which includes nerve cells and their connections located in the rest of the body).
Nicotinic Receptors. Nicotinic receptors exist in skeletal
muscle (the "neuromuscular junction"), autonomic ganglia, the adrenal medulla,
and the central nervous system. Nicotinic receptors consist of five protein
subunits--classically, two alpha subunits, a beta, a gamma, and a delta. (At
the nerve-muscle junction (neuromuscular junction), the alpha subunits are of
type " 1" and an epsilon substitutes for the gamma subunit). Since at least 10
distinct subunit sequences for neuronal nicotinic ACh receptors have
been identified in mammals (2-7, 9,
1" and an epsilon substitutes for the gamma subunit). Since at least 10
distinct subunit sequences for neuronal nicotinic ACh receptors have
been identified in mammals (2-7, 9,  2-4--again, this is as
opposed to muscles, which use the fixed combination
[1]2[1][
2-4--again, this is as
opposed to muscles, which use the fixed combination
[1]2[1][ ][
][ or
or  ] in vertebrates), a great many subunit
combinations are possible. These produce nicotinic receptors with different
structures (though all share the property of having five subunits) and
functional characteristics (Lena and Changeux, 1997). They also have different
affinity for binding to different acetylcholine "agonists" (substances that
bind to acetylcholine receptors and "activate" them) and show different
predilections for desensitization (Corringer, Bertrand, et al., 1998). These
characteristics are of importance in considering possible effects of PB on the
acetylcholine system (see Chapter Thirteen,
"Cholinergic Dysregulation").
] in vertebrates), a great many subunit
combinations are possible. These produce nicotinic receptors with different
structures (though all share the property of having five subunits) and
functional characteristics (Lena and Changeux, 1997). They also have different
affinity for binding to different acetylcholine "agonists" (substances that
bind to acetylcholine receptors and "activate" them) and show different
predilections for desensitization (Corringer, Bertrand, et al., 1998). These
characteristics are of importance in considering possible effects of PB on the
acetylcholine system (see Chapter Thirteen,
"Cholinergic Dysregulation").
Of note, receptors with the 9 subunit are actually stimulated by both nicotine
and muscarine and have atypical "nicotinic/muscarinic" pharmacology (Changeux,
Bessis, et al., 1996).
In each case the subunits are believed to form an "ion channel." Binding of ACh to the receptor produces signaling in the cell by leading to flux of ions through this channel. The different types of nicotinic receptors are not distributed randomly but have characteristic localizations. The nicotinic receptors found at the neuromuscular junction (termed "N1" receptors) are different--in terms of structure and properties--from those at the autonomic ganglia (termed "N2" receptors, which mediate such functions as sweating); they in turn differ from the several varieties found in the CNS. The muscle receptors mediate "fast" effects (as electrical changes in the muscle fiber following ACh binding produce an "action potential," causing the muscle to contract), while central nicotinic effects may have a slower time-course, acting by influencing release of other neurotransmitters, such as dopamine, GABA (gamma amino butyric acid), and glutamate. Although "the nicotinic acetylcholine receptor is the best characterized neurotransmitter receptor" (it has been widely studied in the electric organ of eels and in the easily accessible neuromuscular junction in other animals) (Taylor and Brown, 1994) because of the differences in properties of different nicotinic receptors--and new nicotinic receptors have continued to be characterized--quite a lot remains unknown.
Muscarinic Receptors. Muscarinic receptors are found in the smooth muscle of viscera (internal organs), heart muscle, secretory glands, and in the "endothelial" cells lining blood vessels--as well as in the brain. Muscarinic receptors do not make up ion channels, as nicotinic receptors do. Rather, muscarinic effects are produced more indirectly, through chemicals termed "second messengers." There are at least five distinct muscarinic receptor types (at least five genes have been cloned and sequenced), which differ in their ability to couple to different second messenger "GTP binding proteins," or "G-proteins." These in turn exert other chemical effects, which lead ultimately to muscarinic actions. (The effects of G-proteins are fairly technical and include inhibition of cyclic AMP formation, stimulation of "phospholipase C," and regulation of ion channels--but in this case regulation is not direct, as with nicotinic receptors, but mediated by G-proteins.) (Taylor and Brown, 1994).
Functional Localization of ACh Receptors. As mentioned previously, nicotinic and muscarinic receptors predominate in different places in the body: for example, nicotinic receptors occur in skeletal muscle while muscarinic receptors are found in the smooth muscle of viscera. Moreover, different nicotinic receptors (and different muscarinic receptors) themselves are characteristically found in different parts of the rest of the body--such as "N1" nicotinic receptors at the neuromuscular junction, and "N2" receptors at the autonomic ganglia. Analogously, different receptors dominate in different functions and different parts of the brain. For instance, some areas of the brain (such as the "optic tectum") rely primarily on nicotinic receptors. Some areas--such as the hippocampus (which is critically involved in memory function) and the cerebral cortex--have predominantly muscarinic receptors of the type that bind with high affinity to the chemical "pirenzapine" (which antagonizes the action of ACh and serves as a major factor in distinguishing different types of muscarinic receptors), while the cerebellum and brainstem have predominantly muscarinic receptors that bind this chemical more sluggishly (Taylor and Brown, 1994). As scientists begin to understand more clearly what the effects are of AChE inhibition with such agents as PB on each different receptor type--and begin to understand more clearly which brain effects rely on which receptors--a more satisfactory resolution to the possible contribution of PB to symptoms in PGW veterans may be obtained. This knowledge is still fairly rudimentary.
Terminology Issues. Actions on these receptors lead to characteristic effects, themselves sometimes termed muscarinic, nicotinic, or "central," depending on the type of receptor that produces these effects upon binding by ACh. When this admittedly loose terminology is used, "nicotinic" and "muscarinic" effects refer to effects from action on the peripheral nicotinic and muscarinic receptors. Although central effects themselves result from binding to central nicotinic or muscarinic receptors, it is often not well characterized which receptors are responsible for which effects, so that "central" effects are often referred to. (Nicotine addiction involves central effects that are clearly "nicotinic.") Similar effects--though not identical ones--occur whether increased action of ACh, resulting from inhibition of AChE, is produced by PB, by other carbamates (compounds which, like PB, reversibly "carbamylate" and inactivate the AChE), or by organophosphates (OPs), such as nerve agents or pesticides, which irreversibly "phosphorylate" and inactivate the AChE. A major difference is that since little PB normally crosses the blood-brain barrier into the brain, central effects from PB are normally minimal. In contrast, the related carbamate "physostigmine" does cross into the brain, as do many OPs.
Muscarinic, Nicotinic, and Central Effects of AChE Inhibitors. AChE inhibitors, including both PB and nerve agents, exert their effects by blocking AChE at ACh receptor sites, increasing ACh and ACh activity, causing first hyperactivity of smooth and of skeletal muscles (Medical Letter, 1990) and then paralysis (with high enough doses). If the agent penetrates the CNS, then CNS effects also occur. Nerve agents penetrate the CNS; however, PB does not, or at least not much, when taken under normal conditions. Limited evidence suggests that PB does penetrate the CNS when it is taken under stressful conditions. (See Chapter Seven, "Blood-Brain Barrier Passage.")
Skeletal muscle effects are among the so-called nicotinic effects. Early skeletal muscle symptoms include twitching and cramps and coincide with axonal "backfiring," in which a signal travels up the "axon," the nerve process that relays signals, in the reverse direction to that by which signaling ordinarily occurs. Twitching is first seen in eyelids, and it spreads to face and calves and then becomes generalized. This reaction occurs in the first 24 hours and may be followed by weakness or paralysis, depending on the severity of the intoxication. Paralysis involves all skeletal muscles, including the breathing muscles, and results in labored, shallow, and rapid breathing. Respiratory failure and cyanosis (turning blue) may ensue in some cases. Weakness of the tongue and pharyngeal muscles (muscles in the pharynx, or back of the throat) further enhances respiratory failure by promoting airway obstruction (Gutmann and Besser, 1990; Medical Letter, 1995; Whinnery, 1984).
Muscarinic receptors, as mentioned previously, are found in the smooth muscle of "viscera" (internal organs), heart muscle, secretory glands, and in the endothelial cells lining blood vessels--as well as in the brain. Muscarinic effects from action of ACh at smooth muscles, glands, and the heart include spasm of smooth muscles, increased glandular secretions, and slowing of the heart rate (Hardman, Limbird, et al., 1996). Specific examples of smooth muscle spasm and increased secretions include increased abdominal motility and intestinal secretions, with possible consequent abdominal cramping, nausea, vomiting, and diarrhea; excessive salivation (with drooling); lacrimation (increased secretions from tear ducts); rhinorrhea (runny nose); bronchorrhea (increased bronchial, or airway, secretions); bronchospasm (constriction of the airways); laryngospasm (constriction of the larynx area); diaphoresis (sweating); ureteral spasm with frequent urination or bladder incontinence; and sphincter relaxation, which may promote bowel incontinence. Miosis, or pupillary constriction, is almost always present. In addition, there may be reddening of the eyes and slowing of the heart (Gutmann, and Besser, 1990; Medical Letter, 1995; Whinnery, 1984; Taylor and Brown, 1994). Nicotinic and muscarinic effects on the heart rate are opposed, so theoretically either an increased or reduced heart rate may occur, but bradycardia (slowed heart rate) is more common.
ACh influences autonomic ganglia through both muscarinic and nicotinic mechanisms. Effects also include release of "catecholamines," such as adrenaline (nicotinic). Consequences of these effects may include pallor and transitory elevation of blood pressure followed by low blood pressure (Gunderson, Lehmann, et al., 1992).
So-called central effects result from accumulation of ACh at central synapses. As previously noted, central receptors are themselves characterized as muscarinic or nicotinic, but because it is often not well known which receptors are responsible for which effects, effects are commonly referred to simply as "central." Early signs include anxiety, restlessness, emotional lability, insomnia, and excessive dreaming. (Sleep EEG changes may also be seen--Janowsky, personal communication, 1997.) Larger doses may lead to headaches, tremor, drowsiness, memory impairment, apathy, fatigue, and depression. Severe intoxication results in confusion, ataxia, dysarthria and absent muscle-stretch ("myotatic") reflexes and progresses to coma, Cheyne-Stokes respirations (an abnormal breathing pattern), generalized grand mal-like seizures, and central respiratory depression (breathing problems produced by abnormal action of the breathing center in the brain) (Gutmann and Besser, 1990).
Delayed or chronic CNS effects (Gunderson, Lehmann, et al., 1992) may include giddiness, tension, anxiety, jitteriness, restlessness, emotional lability, excessive dreaming, insomnia, nightmares, headaches, tremor, withdrawal and depression, bursts of slow waves of elevated voltage in EEG (especially on hyperventilation), drowsiness, difficulty concentrating, slowness of recall, confusion, slurred speech, and ataxia (incoordination with walking) (Gunderson, Lehmann, et al., 1992).
Enzymes Regulate ACh Activity. ACh activity is regulated in large part by the enzyme acetylcholinesterase (also called "red blood cell cholinesterase" or "RBC cholinesterase" or "true cholinesterase" and abbreviated "AChE"), which binds to and breaks down ACh that has accumulated in the synapse, preventing inappropriate excessive signaling. Butyrylcholinesterase (also called "plasma cholinesterase" or "pseudocholinesterase," and abbreviated "BChE") plays a less critical role. It is made in the liver and circulates in the plasma. But it is thought that very little ACh will diffuse far from the synapse, so this enzyme plays a small direct role. A possible role for this enzyme in regulating chemicals like PB will be discussed in Chapter Eight ("Individual Differences in Response to PB").
Pharmacokinetics and Pharmacodynamics of PB
Pharmacokinetics refers to the study of the bodily absorption, distribution, metabolism, and excretion of drugs. Pharmacodynamics is a branch of pharmacology dealing with the reactions between drugs and living systems. This section will describe the pharmacokinetics, and to some degree the pharmacodynamics, of PB; these topics are important because they begin to illustrate individual differences in response to PB. Such individual differences could have relevance to symptoms in some PGW veterans.
PB is a positively charged compound that exerts its primary action by inhibiting the enzyme AChE. (It has other actions, including direct binding to, and stimulation of, the ACh receptor.) Because of its charge, PB has difficulty passing through biological membranes, which are relatively impermeant to charged molecules. Perhaps in part for this reason, PB is poorly and erratically absorbed from the gastrointestinal tract into the bloodstream. It has also been suggested that the "low oral bioavailability" of PB may result from "first pass" metabolism by the gut or liver (Leo and Grace, 1996). Recommended oral doses (per the manufacturer) are 30 times those of intravenous doses to achieve similar levels of activity in the body (Williams, 1984), and one author concluded that oral bioavailability amounted to only 7.6 percent--or one-thirteenth--of the administered dose (Aquilonius and Eckernas, 1980). However, other sources state, without citation, that about 40 percent of PB is absorbed following oral administration (Wannarka, 1984), indicating an oral dose would need to be 2.5 times the intravenous dose for the same amount to ultimately reach the blood.
PB makes its way to many sites throughout the body. After radioactive PB is given orally to animals, radioactivity (and, by presumption, PB) is detected in most tissues except the brain, intestinal wall, fat, and thymus (McEvoy, 1991). (PB has low oral bioavailability because of its ionic charge; although a fraction makes its way through the intestinal wall, it does not remain in residence in the intestinal wall.) PB has also been reported to cross the placenta and to decrease fetal plasma cholinesterase activity after large oral doses (McEvoy, 1991).
Most of the orally administered dose is eliminated in the feces (Whinnery, 1984). Most of the absorbed dose (the amount that enters the bloodstream from the GI tract) is eliminated through the kidneys unchanged or as the major metabolite, 3-hydroxy-N-methyl pyridium (3HMP) (Kornfeld, Samuels, et al., 1970; Somani, 1983); other metabolites, including 3-hydroxyphenyltrimethylammonium (3HP) (Hennis, Cronnelly, et al., 1984), are present to a lesser degree (Kornfeld, Samuels, et al., 1970). Liver enzymes appear to play some role in metabolism at least in animals (McEvoy, 1991), particularly the microsomal glucuronidation of 3HMP (Somani, 1997), and there is a two- to threefold increase in concentration of PB in liver microsomes (Somani, 1977; Somani, 1997). According to one source, about 90 percent of the absorbed dose is metabolized by the liver on first pass (Wannarka, 1984) (data not referenced); however, other sources suggest that 75 to 90 percent is excreted unchanged in the urine (Cronnelly, Stanski, et al., 1980; Kornfeld, Samuels, et al., 1970; Keeler, 1990) or that as much as a third of the amount excreted may be the 3HMP metabolite. The 3HMP product is an acetylcholine antagonist (Yanaura, et al., 1993) and also inhibits AChE (Lee, Stelly, et al., 1992). Even after intravenous administration, in which there is no time burden getting the drug into the bloodstream, some PB activity can be detected in the urine 72 hours later (McEvoy, 1991). The half-time of elimination (estimates for the half-life with oral dosage are on the order of two to four hours; see Appendix A) is not stable but lengthens as PB is excreted; this could relate to the putative sequestering of PB in sites such as cartilage (see below "Accumulation of PB in the Body"). Because PB enhances the propulsive movement of the intestines, it may reduce its own absorption with continued use by encouraging its own elimination before the GI tract has had adequate time to absorb the PB.
Patients with severe myasthenia gravis seem to metabolize and excrete PB faster than patients with milder disease, which has been offered as an explanation for the resistance to anticholinesterase medication (that is, to the action of PB) that occurs with some severely ill patients (McEvoy, 1991).
Dosage requirements in myasthenia vary widely due to individual differences in absorption, metabolism, and excretion of PB (as well as differences in disease severity), so that dosage is usually determined individually for each patient. "Many of the same individual variations are probably present in normal subjects" (Whinnery, 1984); Chapter Eight explores whether these variations may be important for ill PGW veterans. PB reportedly has a variable duration of action in patients with myasthenia gravis, supposedly "depending on the physical and emotional stress suffered by the patient and the severity of the disease" (McEvoy, 1991), though other biological factors may be important. Moreover, individual muscle groups in the same patient may respond differently to the same dose of PB, producing weakness in one muscle group while increasing strength in another. The muscles of the neck and of chewing and swallowing are usually the first to be weakened by overdose, followed by the muscles of the shoulder and upper extremities, and finally the pelvic girdle, the muscles that control eye movement, and leg muscles (McEvoy, 1991).
The main "therapeutic" action of PB is AChE inhibition--that is, inhibition of the AChE enzyme, which breaks down ACh. The degree of AChE inhibition that occurs after PB administration is variable. In one small study, in which multiple doses of PB were given (30 mg every eight hours--the same dosing schedule given to PGW personnel deemed to be under threat of chemical warfare attack, but here given for six doses in eight healthy males), AChE inhibition was largely within the target range for nerve agent pretreatment, that is, 20-40 percent inhibition, following the first day of treatment (Sidell, 1990). However, other studies have shown substantial variability in response. In one study, peak inhibition varied from 20 percent to 39 percent of baseline activity, and the period of inhibition exceeding 20 percent varied from one-half hour to five hours--a difference of a factor of 10 (Sidell, 1990). In another study, AChE inhibition after a single dose ranged from 18 to 57 percent (Kolka, Burgoon, et al., 1991b). In a recent larger study of 90 healthy male and female volunteers ages 18 to 44, though ranges were not given, large individual differences were reflected by quite high standard errors in some groups of subjects, particularly for the time of maximum AChE inhibition after continued use (Lasseter and Garg, 1996). While significant differences in AChE inhibition did not occur as a function of sex or weight category in this study, this is likely a reflection of the large individual differences within each group obscuring any between-group differences. (More data regarding variability are discussed in Chapter Eight.)
Because of marked differences in the absorption of PB, the same administered dose may not lead to the same blood level of PB (Cohan, Drettchen, et al., 1977; Marino, Schuster, et al., 1996; Parker, Barber, et al., 1989). Indeed, one study found a more than sevenfold difference in steady-state plasma concentration between patients taking approximately the same daily dose of PB (Aquilonius, Eckernas, et al., 1980). However, the relationship between blood levels of PB and RBC AChE activity is itself not as strong as one might suppose, leading to further variability in response. The correlation between PB and AChE inhibition was only -0.61 in one study (absence of correlation is 0, and a perfect correlation is 1 or -1), though in another the correlation was significant (r = -0.87, p < .05) (Kolka, Burgoon, 1991b). (A correlation of 0.61 implies that only about 36 percent of the variance in AChE inhibition can be explained by blood level of PB.) Moreover, percentage of RBC cholinesterase (acetylcholinesterase) inhibition was not highly correlated with weight, height, or body surface area (Kolka, Burgoon, et al., 1991b). Thus, in addition to marked variability in absorption of PB, the blood levels of PB following absorption do not necessarily predict the extent of AChE inhibition. Thus, variation in AChE inhibition following PB administration appears to be due to individual differences in amount of drug absorbed, in rate of elimination of the drug, and in sensitivity of AChE to inhibition by PB (Sidell, 1990). Later, evidence will be shown that AChE inhibition in turn does not strongly predict toxicity. Table 3.1 shows the several identified factors (in the pathway from administration of PB to response) which may lead to differences in individual response to PB for the same oral dose of PB.
| Similar Exposure | Different Response |
| Same oral dose | Different amount entering blood (differences in absorption, peristalsis) |
| Same PB entering blood | Different levels of PB in blood over time (differences in clearance/metabolism) |
| Same PB in blood | Different levels of AChE inhibition |
| Same AChE inhibition | Different effect/toxicity |
Accumulation of PB in the Body. Some research has found that rats subcutaneously administered radioactive PB twice daily for 16 days do not excrete 100 percent of the daily dose of PB. On average, 76 percent was excreted as PB and its metabolites in urine and 7 percent in feces, leaving 17 percent unaccounted for, suggesting accumulation of the drug and its metabolites in the body with multiple dosing. Progressive increases in radioactivity per gram of tissue were also noted, and it was suggested that PB may bind specifically to chondroitin sulfate, since the radioactivity accumulated strongly in the ear and tail, which are cartilaginous tissues (Somani, 1977; Somani, 1983; Somani, 1997).
Pharmacokinetic and Pharmacodynamic Data: Extrapolation from Myasthenics to Normals. Some maintain that pharmacokinetic and pharmaco-dynamic results, as well as toxicity information for healthy subjects and patients with myasthenia gravis, are similar, and the data from myasthenia gravis should be essentially applicable to all individuals (Whinnery, 1984). However, there are several reasons to be concerned that data from myasthenics and normals may differ (see Table 3.2).
| Myasthenia | Normal |
| PB restores nicotinic activity toward normal at the muscles | PB raises nicotinic activity away from normal |
| Accelerated metabolism? | Metabolism not accelerated |
| Use is continued indefinitely | Use is terminated |
First, high doses of PB are given to myasthenics in order to raise their neuromuscular ACh activity toward normal. However, high doses of PB in nonmyasthenics would raise neuromuscular ACh to supranormal--away from normal. Thus, it cannot be assumed that PB's safety for myasthenics would be the same for those without myasthenia--particularly with regard to nicotinic effects, the chief source of severe side effects. (Indeed, as long ago as 1984 it was observed that "the amount of pyridostigmine which can be administered without severe side effects appears to be related to the degree of impairment of neuromuscular transmission" (Williams, 1984).) With regard to muscarinic side effects, these are often experienced by patients with myasthenia gravis (Hood, 1990); indeed, in one study, the reported side effect rate in myasthenics was 34 percent (Beekman, Kuks, et al., 1997). One analogy is the difference between how insulin affects diabetics and those not suffering from the condition. A severe diabetic may tolerate or require 60 units of insulin or more each day to bring his or her blood sugar toward normal. Yet far smaller doses may induce hypoglycemic coma or even death in a nondiabetic subject. Similarly, a high dose of PB may be harmless or indeed salutary in a patient with low acetylcholinergic activity (e.g., myasthenia gravis), but even a lower dose may be frankly harmful in an individual with normal ACh activity.
Second, patients with severe myasthenia appear to metabolize and excrete PB faster than those with milder disease (McEvoy, 1991). By extrapolation, myasthenics may have a heightened metabolism of PB compared to normals (perhaps resulting from alterations in metabolism with regular use rather than because of the disease per se). For these reasons, and others presented at the start of Chapter Fourteen (explaining why cessation of PB may pose problems that will not be manifest in lifelong users like patients with myasthenia gravis), data from patients with myasthenia--or perhaps from those who use PB regularly--may not apply to those naive to PB.
A third reason that data from myasthenia may not apply relates to the fact that patients with myasthenia use PB indefinitely, while Gulf War use was transient. Some evidence suggests that use of PB may lead to downregulation of the acetylcholinergic system (see sections on Downregulation and Motor Adapter Effects). Use of PB and other AChE inhibitors in animals has been shown to cause reduced ACh release, withdrawal of nerve fibers from junctional folds and reduced sensitivity of ACh receptors--effects that would be particularly offset by continued PB use (and indeed were generated by the body in an effort to restore regulation in the face of PB) but that might generate problems with PB discontinuation. The duration of these effects is not well characterized.
Additional differences in sensitivity to PB may result from the slow up-titration of PB in myasthenics. Those who have been brought to high doses may have heightened muscarinic activity (as a result of the higher dose of PB, since myasthenics do not have impairment in muscarinic cholinergic activity but only in nicotinic activity). This may result in faster peristalsis leading to reduced absorption of PB. For whatever combination of reasons related to absorption and elimination, it has been noted that myasthenics may develop a tolerance to, or loss of clinical response to PB (McEvoy, 1991). For these reasons, caution is warranted in extrapolating pharmacokinetic data, and evidence on side effects, from patients with myasthenia to nonmyasthenics. (This issue is discussed again in Chapter Fourteen, "Chronic Effects"; background from intervening chapters will allow additional material to be brought to bear.)
Other Actions on the Cholinergic System
Partial Agonist. PB is considered an AChE inhibitor. However, studies in diverse species (including amphibians, fish, and mammals) indicate that PB also reacts directly with the ACh receptor as a "partial agonist"; it produces some of the same effects that ACh itself would produce acting at the same receptor (Aracava, Deshpande, et al., 1987). Moreover, ion channel openings, which are normally stimulated by nerve signals that result in ACh binding to the receptor, show increased noise (Aracava, Deshpande, et al., 1987), which could theoretically adversely affect information-processing capabilities. (Opening and closing of ion channels is the means by which nerve signals are typically propagated.)
ACh Receptor Desensitization. Moreover, PB either alone or in combination with ACh induces an altered, "desensitized" species of the nicotinic receptor-ion channel complex (Aracava, Deshpande, et al., 1987). That is to say, PB use produces ACh receptors (binding sites) that are less sensitive to the action of ACh. (This has been proposed as one of the methods of conferring protection against nerve agent.) Normally, when ACh binds, certain ion channels open and produce an electrochemical signal. The frequency of these channel openings is decreased when PB is given, and ACh ion channel "currents" are modified (Sherby, Eldefrawi, et al., 1985; Akaike, Ikeda, et al., 1984). Other carbamates (chemicals in the same family as PB) produce similar but not identical effects (Sherby, Eldefrawi, et al., 1985; Shaw, Aracava, et al., 1985). This desensitization could have a role in cholinergic downregulation (see Chapter Thirteen).
Autoregulation. Most ACh receptors considered clinically are those that exist at the "postsynaptic" membrane--the membrane of a signal-receiving cell. ACh released from a signaling or "presynaptic" cell (a nerve cell or "neuron") binds to these receptors on the postsynaptic cell (typically either a nerve or muscle cell), allowing the presynaptic cell to communicate with the receiving cell. However, ACh receptors also exist at the presynaptic membrane--the membrane of the signaling cell--where they play an important role in "autoregulation" of ACh release from the terminal axon (the extension of the signaling cell that grows out and forms connections with the receiving cell). When ACh binds to and activates these presynaptic receptors, the effect is to inhibit further release of ACh from the presynaptic membrane (that is, from the signaling cell) (Gutmann and Besser, 1990). (However, nicotinic action presynaptically increases release in the brain of other neurotransmitters, including both glutamate and GABA.) The factors that regulate the degree to which this inhibition occurs are not understood. Thus, inhibition of ACh breakdown by PB will lead to increased binding by ACh on these presynaptic sites, potentially reducing continued release of ACh. No data were found to elucidate the strength or time-course of this autoregulatory effect.
ACh Receptor Heterogeneity. Neuronal ACh receptors exhibit "considerable heterogeneity with respect to their distribution, pharmacological sensitivity and functional role" (Albuquerque, Costa, et al., 1991). Many receptor types have been identified in the CNS (see the Chapter Thirteen discussion of neurotransmitter dysregulation); since these have different characteristics, they are presumably differently affected by such carbamates as PB, in the event that PB reaches the CNS.
Nonsynaptic Cholinergic Effects. It has been noted that "the presence of acetylcholine and its receptors at locations not related to synaptic activity raises the possibility that acetylcholine may have receptor-mediated cellular functions other than transmission of nerve signals, i.e., that acetylcholine may act as a local hormone modulating cellular functions" (Grando, Horton, et al., 1995). For example, both nicotinic and muscarinic ACh receptors, similar to those seen in the brain and autonomic ganglia, evidently regulate migration and cell stickiness of skin cells (epidermal keratinocytes). Immune and blood cells also have cholinergic mechanisms, as do sperm and ovaries (Schwarz, Glick et al., 1995).
Effects on Other Transmitter Systems
ACh (nicotinic) stimulation can cause release of neurotransmitters including GABA, dopamine, and glutamate (Kayadjanian, Retaux, et al., 1994; McGehee, Heath, et al., 1995; Role and Berg, 1996). Moreover, there appears to be a relation to serotonin action (Albuquerque, Pereira, et al., 1997a; Albuquerque, Pereira, et al., 1997b). It has been speculated that in addition to the actions of ACh in the usual signaling-cell/receiving-cell interactions (the "wiring" transmission mode), ACh may also be released in a "paracrine" or "volume transmission" fashion--acting more or less like a local hormone, drifting to modulate release of other neurotransmitters (not necessarily just at a site at which the ACh-releasing cell synapses) (Agnati, Zoli, et al., 1995; Changeux, Bessis, et al., 1996). Evidence in favor of this is that the "cell bodies" of the neurons that release ACh are restricted to a few nuclei located mainly in parts of the brain termed the basal telencephalon and dorsal pons, but the ends of the nerve cell processes from these cells are spread throughout the brain in a somewhat diffuse way, and no consistent match exists between the known distribution of the nicotinic ACh receptors in the brain and that of the acetylcholinergic "terminals" (release sites from the signaling cell) (Agnati, Zoli, et al., 1995; Changeux, Bessis, et al., 1996; Role and Berg, 1996). Further complicating matters, muscarinic receptors are known to regulate synaptic transmission in some instances with actions opposite to nicotinic receptors (Changeux, Bessis, et al., 1996), so that the effects depend on the relative distribution of these types of receptors. This distribution, however, has not yet been established (Changeux, Bessis et al., 1996). The information provided below should be considered with these provisos in mind.
As noted elsewhere (see Chapter Fifteen, Other Considerations, Hormone and Stress Effects), ACh may also stimulate release of catecholamines--epinephrine particularly, and also norepinephrine--and hormones, including cortisol, prolactin, growth hormone, beta-endorphin, and others.
Glutamate. The "glutamatergic" (i.e., glutamate-related) transmitter system shares elements with the acetylcholinergic system, including its ability to be influenced by carbamates (such as PB). In the mammalian central nervous systems, the NMDA (N-methyl-D-aspartate) type of glutamatergic receptor as well as the ACh receptor are involved in a number of processes. These range from control of transmitter release (described above), to postsynaptic membrane depolarization (changes occurring in the membrane of the signal-receiving cell) with mobilization of intracellular second messengers. Second messengers, such as calcium, in turn participate in mechanisms of protein synthesis, learning (through a process termed "long-term potentiation"), outgrowth of nerve processes to allow connections to other cells, cell death, and other functions (Albuquerque, Costa, et al., 1991). Glutamate receptor overactivation can induce delayed neuronal cell death (toxic effects by overactivation are termed "excitotoxicity"). Such effects may be mediated by breaking of DNA strands (Didier, Bursztajn, et al., 1996).
Indeed, it has been shown that low (nanomolar) concentrations of nicotine enhance both glutamatergic and acetylcholinergic synaptic transmission by activating presynaptic nicotinic ACh receptors that increase presynaptic calcium concentrations (McGehee, Heath, et al., 1995).
Studies have demonstrated that carbamates and OPs (including the OP agents diisopropylfluorophosphate (DFP) and the nerve agent VX) affect properties of the glutamate system, including such presynaptic properties (properties of the signal-sending side) as increasing transmitter release and such postsynaptic properties (properties of the signal-receiving side) as reducing the peak size of the current induced by binding of glutamate to its receptor. Moreover, carbamates may cause unstimulated endplate synaptic potentials, or voltage changes in the receiving cell side, large enough to provoke "action potentials" (full self-propagating signals down a nerve process that are normally produced in response to input from one or more signaling cells). This response could disrupt the specificity of signaling, since action potentials are more typically driven by specific inputs to the neuron. These data derive from work on insects' neuromuscular junctions, however, and insects employ glutamate rather than ACh at their neuromuscular junctions (Albuquerque, Deshpande, et al., 1985; Aracava, Deshpande, et al., 1987; Idriss, Aguayo, et al., 1986; Albuquerque, 1985). The degree to which PB per se affects glutamate neurons in the mammalian CNS remains to be evaluated but would likely depend in part on the degree to which PB has access to the brain. (It has been observed that ACh receptors and NMDA-sensitive receptors on central neurons share much functional homology with ACh receptors in muscle fibers (Albuquerque, 1988), suggesting that similar effects may apply if PB were to reach the CNS.)
GABA. Carbamates may also affect signaling through another neurotransmitter system, the GABA (gamma amino butyric acid) system. GABA is the chief inhibitory neurotransmitter in the mammalian nervous system (Hardman, Limber, et al., 1996). ACh acts presynaptically to modulate GABA release. Preterminal nicotinic receptors on GABAergic axons have been identified in rats (e.g., the interpeduncular nucleus (Lena, Changeux, et al., 1993)), and ACh has been shown to modulate the release of GABA (Albuquerque, Pereira, et al., 1995; Mukhopadhyay and Poddar, 1995; Kayadjanian, Retaux, et al., 1994).
Dopamine. By a similar mechanism, ACh acts via presynaptic nicotinic
receptors to modulate the release of dopamine (Albuquerque, Pereira, et al.,
1995; Kayadjanian, Retaux, et al., 1994). Indeed, in some parts of the brain
(but not others), the release of GABA by nicotine is mediated by dopamine
release (Kayadjanian, Retaux, et al., 1994). Nicotine evokes dopamine release
(from the rat corpus striatum as well as frontal cortex) in vivo and in vitro.
Chronic treatment with nicotine has been shown to increase the density of
nicotine-binding sites (that is, nicotinic receptors) in the frontal cortex but
to produce no increase in striatal preparations (Wonnacott, Marshal, et al.,
1994). A reduction in dopamine release is evoked by the same amount of
nicotine in the frontal cortex but not in the striatum (Wonnacott, Marshal, et
al., 1994). There are also antidopamine muscarinic effects (Georguieff,
Lefloch, et al., 1976)--and perhaps wholly or partly because of different
distribution of muscarinic and nicotinic receptors, ACh stimulation regulation
of dopamine activity may be opposite in different brain regions. (For
instance, it is opposite in the "nigral" and "striatal" regions of the brain
(Javoy, Agid, et al., 1974; Nose and Takemoto, 1974; Westernick and Korf, 1975;
Davis, Faull, et al., 1979).) The clinical relevance of an effect of ACh on
dopamine is suggested by reports of development and of exacerbation of
parkinsonism (a condition of reduced motor movement combined with tremor that
results from problems in the dopamine-rich nigrostriatal system in the brain
(Wilson, Braunwald, et al., 1991)), following PB (Iwasaki, Wakata, et al.,
1988) (Kao, Kwan, et al., 1993) and OPs (Davis, Yesavage et al., 1978) or
unspecified pesticides (Butterfield, Valanis, et al., 1993; Hubble, Cao, et
al., 1993)). Also, the 2-containing neuronal nicotinic ACh receptor is
believed to assist in mediating the reinforcing properties of nicotine (and
other substances of abuse), by contributing to release of dopamine in the
"mesolimbic" system of the brain (Picciotto, Zoli, et al., 1998).
Substance P. The cholinergic system, specifically AChE--and by extension PB--affects breakdown of "substance P" (Chubb, Hodgson, et al., 1980), a neurotransmitter involved in pain signaling.
Serotonin. Serotonin is known to be involved in important functions including regulation of mood, pain, impulsivity, and sleep. (See section on Sleep and Serotonin in Chapter Fifteen, "Other Considerations.") Of note: Several lines of evidence suggest important interactions between the ACh system and the serotonergic system. (A later section in Chapter Fifteen discusses considerations of illnesses in PGW veterans as they may relate to serotonergic dysfunction.)
Briefly, PB binds to the same site on the ACh receptor that serotonin is believed to bind to, a relatively new finding (Albuquerque, Pereira, et al., 1997a; Albuquerque, Pereira, et al., 1997b). (This site has also been called the "galanthamine binding site.") Although many identified classes of nicotinic receptors are in the brain that differ by which of a set of subunits make up their five parts, this binding site is highly "conserved" across different types of nicotinic receptors--that is, while other properties of the receptor change, this property remains constant. Carbamate and OP AChE inhibitors, including nerve agents and PB, have been shown to reduce platelet serotonin uptake, increase serotonin turnover, and reduce nighttime enhancement of serotonin in the pineal gland, and nighttime rise of melatonin measured in blood. Some serotonergic drugs have been found to augment the AChE-inhibiting effects of PB and slow the time-course of recovery. Others have been shown to have similar effects to PB in some circumstances, such as enhancing release of growth hormone from the pituitary gland in response to growth hormone releasing hormone from the hypothalamus. Finally, there has been an unsubstantiated (non-peer reviewed) report of benefit from combined serotonergic/dopaminergic treatment of ill PGW veterans (Hitzig, 1997). This treatment used the drug combination fenfluramine and phentermine. (Fenfluramine is no longer available, after reports of heart valve abnormalities with this drug or with the fenfluramine-phentermine drug combination.) Some issues related to serotonin are discussed in greater detail in Appendix B.
Opioids
The opioid codeine has, like serotonin, been shown to bind to the "galanthamine" binding site on nicotinic ACh receptors, acting as a modulation of central nicotinic function (Albuquerque, Pereira, et al., 1997b). (It is known that the opioid system, the serotonin system, and the acetylcholinergic system are all involved in regulation of pain. High levels of opioids and serotonin are related to increased tolerance to pain. More on the relationship between cholinergic function and pain will be discussed in Chapter Fifteen, in the section on "Sleep.")
Shelf Life
PB can be maintained for long periods without refrigeration (FDA, 1997). Because of the desire for long storage, the military policy is to maintain PB under refrigeration and dispose of PB that has remained unrefrigerated for a period exceeding six months. Refrigeration of PB is desired because prolonged storage without refrigeration may affect its potency. Discussion with FDA representatives indicates that PB does not break down into toxic products (FDA, 1997).
Breakdown Products
As noted above, the chief metabolite of PB is 3-hydroxy-N-methylpyridium. This product is discussed earlier in the section on pharmacokinetics and pharmacodynamics. Although it has traditionally not been thought to contribute to the action of PB, it may have some AChE inhibiting activity (Lee, Stelly, et al., 1992).
PB was licensed by the FDA in 1955 for use in patients with myasthenia gravis and to reverse effects of nondepolarizing neuromuscular blockers (De Fraites, 1996).
Myasthenia Gravis
Myasthenia gravis is a disease characterized by increased weakness and fatigability of muscles caused by an autoimmune attack on ACh receptors at the junction of nerves and muscles. Antibodies are produced by the body to nicotinic ACh receptors at the neuromuscular junction (the junction at which the nerve joins the muscle and can signal the muscle to contract). These receptor antibodies attack the receptors and lead to the loss of functional receptors at which ACh can act. It is the action of ACh at the muscle's "motor endplate" that causes muscle fibers to contract; loss of the ability to contract produces muscular weakness (Drachman, 1994). Myasthenia gravis has a prevalence of 50 to 169 cases per million population, with approximately 25,000 affected persons in the United States (Antonini, Morino, et al., 1996; Drachman, 1994). The annual incidence is in the range of 2-10 per million population (Schon, Drayson, et al., 1996; Somnier 1996). It has a bimodal age distribution, with a primarily female early peak in the second and third decades and a late peak affecting predominantly males from 60 to 74 years old (Drachman, 1994; Antonini 1996, Morino, et al., 1996). Ten to 20 percent of patients do not have detectable antibodies, but it is believed that circulating antibodies are present even in these individuals, since passive transfer of immunoglobulins (antibodies) from these patients to mice leads to loss of ACh receptors at the mouse neuromuscular junction (Drachman, 1994). Experimental myasthenia has been developed in mice, in which there are antibodies to ACh receptors (Berman and Patrick, 1980).
PB is used as a treatment for myasthenia gravis. Myasthenics' loss of ACh receptors results in reduced ACh binding to receptors, which in turn results in reduced "cholinergic" activity. PB treatment increases the availability of ACh to bind remaining receptors (Drachman, 1994), partially restoring cholinergic activity. The maximal useful dosage rarely exceeds 120 mg each three hours; higher doses may increase weakness. Benefit is usually incomplete and often wanes after weeks or months of treatment (Drachman, 1994). Increasing doses are often needed, and patients may become unresponsive to PB after prolonged treatment (McEvoy, 1991). In some instances, responsiveness may be restored by reducing the dose or withdrawing the drug for several days under medical supervision (McEvoy, 1991).
Variant myasthenic syndromes with different mechanisms exist and patients with these syndromes respond poorly to PB (Engel, Lambert, et al., 1977, 1981). These syndromes are uncommon.
Reversal of Neuromuscular Blockade
PB is used to reverse a neuromuscular blockade (used in anesthesia) by "nondepolarizing neuromuscular blockers" (such as tubocurarine, metocurine, gallamine, pancuronium) following surgery, in intravenous doses ranging from 2 mg to 18 mg (Alisoglu, Nagelhout, et. al., 1995; McNall, Wolfson, et al., 1969). PB is not useful for reversing the action of "depolarizing" neuromuscular blockers (such as succinylcholine or decamethonium) and may actually prolong the action of these agents (McEvoy, 1991; Fleming, Macres, et al., 1996). This role for PB is relevant only in that it is another circumstance in which PB has been approved for use by the FDA and has been used with apparent safety. Its use in this circumstance, as in the instance of myasthenia (but in contrast to its use as a nerve agent pretreatment adjunct), also serves to bring muscle activity toward normal.
Treatment of Fatigue Due to Other Causes
It has been noted that carbamates (like PB) can temporarily increase clinical strength even for normal individuals (Rustam, Von Burg, et al., 1975). PB has been used with some reported success in the treatment of fatigue in nonmyasthenic conditions.
PB has been used in treating the fatigue of patients with post-polio syndrome, in which presynaptic defects (defects in signal sending, rather than postsynaptic defects in signal receiving, as in myasthenia gravis) are thought to be present (Trojan and Cashman, 1995a; Trojan and Cashman, 1995b). The benefit may result from increased strength rather than reduced fatigability. Post-polio syndrome patients appear to have symptoms of both central and peripheral fatigue (Trojan and Cashman, 1995a).
PB has been found to benefit symptoms and quality of life in HIV-positive patients who test positive for ACh receptor antibodies (in open-label trials using 60 mg three times daily or four times daily), though the role of antibodies in these patients' symptoms is unknown (Cupler, Otero, et al.,1996).
PB has also been used for treatment of "drop attacks" in the elderly; elderly persons with drop attacks uniformly reported relief from falls with PB (Braham, 1994). It has been postulated that the postural righting reflex in these individuals requires a prompt neuromuscular response, which is aided by PB. Other individuals, including supposedly "neurotic" subjects with subjective weakness or fatigue in whom tests for ACh receptor autoantibodies are negative, have experienced relief with use of PB (Park, Kim, et al., 1993).
Neostigmine, a relative of PB, has been used with success in some Iraqi patients suffering from methylmercury poisoning. Use of this agent led to doubling of strength and large fluctuations in strength with administration and withdrawal of the drug, employing doses that might produce signs of toxicity in normal individuals (Rustam, Von Burg, et al., 1975). Again it seems that subjects with a clinical need for PB tolerate higher doses without apparent adverse effect than do "normal" individuals.
(Of note, other AChE inhibitors that penetrate the brain, such as tacrine, have been used to reduce symptoms of dementia in the elderly.)
Dose
In myasthenia gravis, dosing is usually initiated at 60 mg three times daily (twice the PGW dose), with dosage increased gradually until no further benefit to strength is seen (McEvoy, 1991). The average daily dose is 600 mg/d orally (60 mg 10 times per day, or about 7 times the PGW dose), with an acceptable dosage range of 360 to 1500 mg/d (up to ~17 times the PGW dose) (McEvoy, 1991). The literature reveals a de facto dose range from 30 to 2,000 mg/d, with up to 6,000 mg/d recorded in severe cases (~67 times the PGW dose) (Whinnery, 1984). In the reversal of a neuromuscular blockade, doses from 0.1 to 0.25 mg/kg, or 4 to 20 mg, have been quoted. These doses are quite different from those given to myasthenics in part because poor absorption of orally administered drug (in myasthenia) requires high doses compared to drug administered by injection (in reversal of neuromuscular blockade).
Possible Adverse Effects of PB Treatment in Myasthenia Gravis
Some concerns have been raised regarding possible deleterious effects of PB in the treatment of patients with myasthenia gravis, on the grounds that a decrease in the number of patients achieving complete remission after surgical thymectomy has occurred since 1961 (Scadding, Havard, et al., 1985). This effect was thought to be related to use of anticholinesterase therapy. It has been speculated that PB may cause damage to the neuromuscular junction and that this damage--unlike the immune effects of the myasthenia itself--cannot be corrected by surgical removal of the thymus. Symptoms of this damage may be difficult to distinguish from the effects of myasthenia per se (Rockefeller Report, 1997). (See Chapter Twelve, "Neuromuscular Junction Effects.")
PB is used as a "pretreatment" or, more precisely, as a "pretreatment adjunct" for poisoning by the nerve agent soman. It confers no protection on its own but enhances the protection conferred by postexposure treatment in the form of atropine and pralidoxime (2-PAM), when tested in animals.
Dose
Research in the United Kingdom has indicated that AChE inhibition of 20 to 40 percent should be maintained to provide "adequate" protection against death from soman poisoning; generally, 30 mg each eight hours orally has been found to provide that range of inhibition in people (Gall, 1981), although the inhibition may fall below 20 percent six hours after dosing. Dosing every eight hours has been estimated to leave five hours a day in which AChE inhibition falls below 20 percent (Whinnery, 1984). Nonetheless, the recommended dosing schedule for nerve agent pretreatment is 30 mg orally every eight hours.
Duration of Military Use
PB has been stockpiled by the U.S. military since 1986 (Dunn and Sidell, 1989), based on British data, for adjunctive nerve agent pretreatment protection. Though PB is approved by the FDA for use for myasthenia gravis, it remains an investigational new drug (IND) for nerve agent pretreatment by the military. (For more information on this issue, see Rettig, 1999). As noted previously, evidence from animal studies indicates that PB protects against lethality from the nerve agent soman (particularly in primates) and does not interfere in a meaningful way with protection against lethality from other nerve agents (studies done in rodents; no direct evidence in primates). Human studies demonstrating efficacy against nerve agent lethality would be clearly unethical, and this failure to demonstrate efficacy for use in humans--rather than safety concerns--had been the major stumbling block in the licensure of PB for nerve agent pretreatment. Thus, although PB has been stockpiled by the military and the decision was made to use PB pretreatment in the Persian Gulf, PB has not been licensed by the FDA for nerve agent pretreatment. The FDA granted a waiver for the requirement of informed consent for this drug prior to its use in the PGW. For more detail on the history of the U.S. use of PB in the military, and the respective positions of the FDA and DoD on this issue, see Rettig (1999).
What Are Nerve Agents?
Nerve agents include GA (tabun), GB (sarin), GD (soman), GF (cyclosarin), thiosarin, and VX (see Figure 3.2). These agents act by "irreversibly" (i.e., with a very long time-course) inhibiting the action of the enzyme AChE, which is involved in regulating (by breaking down) the neurotransmitter ACh. The resulting inhibition of AChE leads to a buildup of ACh at the synapse or at the motor endplate, the sites at which signaling between nerve cells, or between nerve and muscle, occur. This leads to increased action of ACh, discussed previously as nicotinic, muscarinic, and central effects. (For more information on CW agents, see Augerson, forthcoming.)
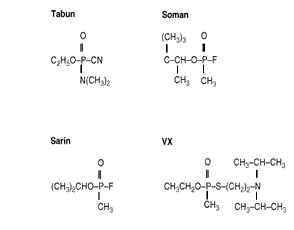
Figure 3.2--Structure of Nerve Agents
What Is the Effect of Postexposure Treatment for Nerve Agents?
Atropine, a muscarinic blocker, antagonizes the muscarinic effects of PB (see "Chemical Characteristics," above). 2-PAM (pralidoxime) enhances the protection by pulling the nerve agent off AChE. Diazepam (Valium) may also be given by injection to prevent CNS effects, particularly seizures, and the long-term cognitive consequences that may ensue from prolonged seizure activity.
Atropine and pralidoxime were given to all troops in a "Mark I Nerve Agent Antidote" autoinjector kit (FORSCOM, 1990). Soldiers carried three 2 mg autoinjectors of atropine for use when symptoms first appeared and three 600 mg autoinjectors of 2-PAM for concurrent intramuscular use with atropine (Gunderson, Lehmann, et al., 1992). Military personnel at risk also carried a 10 mg intramuscular autoinjector of diazepam to administer to their companions if needed (Gunderson, Lehmann, et al., 1992).
Why Is PB Needed in Addition? Pralidoxime ceases to be effective once the effect called "aging" takes place (see Figure 3.3). Aging involves the release of a "leaving group" from the central phosphorus atom of the nerve agent in the nerve agent-AChE complex that permanently inactivates the AChE enzyme (Dunn and Sidell, 1989). (More specifically, there is "partial dealkylation" of the phosphorylated serine group at the active site in the enzyme (Mason, Waine, et al., 1993).)
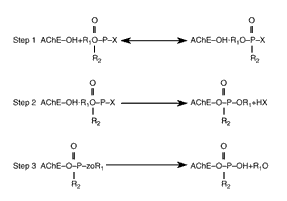
Figure 3.3--Reaction Between Nerve Agent Compounds (or Other OPs) and AChE
Step 1: Formation of AChE-nerve agent complex. Step 2: Phosphorylation and inactivation of the enzyme. Step 3: Aging reaction, in which a "leaving group" separates, providing a monophosphoric acid residue bound to the enzyme. Following the aging process, the nerve agent can no longer be pulled off the AChE molecule by pralidoxime (adapted from Sidell and Borak, 1992).
For those exposed to sarin, there is ample time following exposure to administer an oxime before aging of the nerve agent-AChE complex occurs. Sarin's half time to aging (time at which aging has occurred in half the sarin-AChE complexes) is several hours. The aging half-time is even longer for tabun, cyclosarin, and VX (Table 3.3). However, the half-time of aging for soman is approximately two minutes, inadequate for even highly trained personnel to administer atropine and oxime.
| Nerve Agent | Aging Half-Time | Source | Reference |
| Soman (GD) | 2�6 1 min 8 min 9 min |
Human, in vitro Marmoset, in vivo Guinea pig, in vivo Rat, in vivo |
Mager, 1984 Talbot, Anderson, et al., 1988 Talbot, Anderson, et al., 1988 Talbot, Anderson, et al., 1988 |
| Sarin (GB) | 5 h 3 h |
Human, in vivo Human, in vitro |
Sidell and Groff, 1974 Mager, 1984 |
| Cyclosarin (GF) | 8 h 40 h |
Human, in vitro Human, in vitro |
Hill and Thomas, 1969 Mager, 1984 |
| Tabun (GA) | 13 h 14 h |
Human, in vitro Human, in vitro |
Doctor, Blick, et al., 1993 Mager, 1984 |
| VX | 48 h | Human, in vivo | Sidell and Groff, 1974 |
NOTE: Adapted from Dunn, et al., 1997, and New Drug Application to FDA for PB (Pyridostigmine, 1996; Dunn, Hackley, et al., 1997).
It has been estimated that, for adequate protection against nerve agents in combat conditions, a protective ratio of at least 5 is desired (Dunn, Hackley, et al., 1997; Dunn and Sidell, 1989). That is, the treatments we give must allow exposure to five times the usual lethal dose. The protective ratio (PR) is the LD50 with treatment (the dose that is lethal in 50 percent of subjects, if treatment is given) divided by the LD50 without treatment (the dose that is lethal in 50 percent if treatment is not given) (Sidell, 1990). In the absence of PB, the PR of atropine and oxime against the nerve agent soman does not reach this goal but is at best around 1.6 in primates (Dunn and Sidell, 1989). However, adding PB brings the ratio to in excess of 5 in primates (Dunn and Sidell, 1989), providing more adequate protection against death from soman (Table 3.4).
| 10 LD50 | 5 LD50 | 1 LD50 | |
| Soman (GD) | |||
| Liquid artillery |  43 43 |
22 |
3 |
| Liquid bombs | 65 |
62 |
56 |
| Vapor artillery | 90 |
86 |
77 |
| Vapor bombs | 78 |
70 |
63 |
| Sarin (GB) | |||
| Liquid artillery | 40 |
15 |
1 |
| Liquid bombs | 24 |
15 |
7 |
| Vapor artillery | 100 |
100 |
90 |
| Vapor bombs | 99 |
92 |
72 |
| Cyclosarin (GF) | |||
| Liquid artillery | 43 |
21 |
4 |
| Liquid bombs | 67 |
60 |
54 |
| Vapor artillery | 91 |
85 |
75 |
| Vapor bombs | 77 |
72 |
63 |
| Tabun (GA) | |||
| Liquid artillery | 70 |
50 | 5 |
| Liquid bombs | 70 |
62 | 57 |
| Vapor artillery | 100 |
95 | 84 |
| Vapor bombs | 90 |
84 | 69 |
| Vx | |||
| Liquid artillery | 70 |
47 |
14 |
| Liquid bombs | 40 |
25 |
14 |
| Vapor artillery | 2 |
2 |
1 |
| Vapor bombs | 4 |
4 |
3 |
A PR of 5 was chosen as the target PR based on such findings as those in Table 3.4, taken from the New Drug Application for PB (Pyridostigmine, 1996). This shows results of computer models of possible battlefield scenarios based on U.S. weapons engineering that were used to estimate the level of exposure of troops to nerve agents in various combat situations. It indicates the percentage of the population within the "target area" that would be exposed to various levels of nerve agent. A "standard" target area for analysis for an artillery unit is 600 meters by 400 meters (Pyridostigmine, 1996). For soman (GD), with liquid exposure through either artillery or bombs, or with vapor exposure from bombs, a majority would be exposed to fewer than 5 LD50s of nerve agent; and 7 percent or fewer would be exposed to more than 5 LD50s but fewer than 10 LD50s. That is, most of the people who would be protected if a PR of 10 could be obtained would also be protected by a PR of 5. For some personnel with higher nerve agent exposures, such as personnel near "ground zero" (the point of munition impact), CW exposure may not constitute the most serious injury.[1]
How Does PB Confer Additional Protection? PB enhances the protection of oxime and atropine against a nerve agent, perhaps by reversibly binding to the AChE site at which the nerve agent or other cholinesterase inhibitors (like the OP DFP) would normally bind (Koelle, 1946; Sidell, 1990), preventing the nerve agent from binding. In particular, soman cannot bind to a PB-bound AChE molecule. This prevents aging of soman-AChE from taking place. (Without PB, this would lead to irreversible inhibition of the soman-bound AChE molecules, and recovery of AChE activity would require several weeks for full regeneration of AChE.) The stated goal of PB pretreatment is to bind (and thereby inhibit) approximately 30 percent of AChE; in animal studies, carbamylation of about 30 percent of circulating (RBC, or erythrocyte) AChE corresponds to a great increase in the effectiveness of the antidotes (Sidell, 1990). This 30 percent of AChE will be "protected" from binding and irreversible inhibition by soman and will be restored to normal function once spontaneous "decarbamylation" (dissociation of PB from the AChE molecule) occurs. (Meanwhile, there is enough time for oxime to be given, so that PB decarbamylates, and any residual nerve agent that binds the now-functional AChE can be pulled off.) In animal studies, however, a relationship between the rate of reactivation of AChE inhibited by a pretreatment compound (or the level of RBC AChE inhibition) and the protection against soman has not always been evident (Langenberg, De Jong, et al., 1996; Jones, Carter, et al., 1985), leading to some doubts regarding whether this mechanism adequately explains protection against soman (Prendergast, 1997).
Other mechanisms have been proposed that may contribute to protection by PB pretreatment, including desensitization of the neuromuscular endplate and ACh receptor; decreased quantal release of ACh; and decreased frequency of "miniature endplate potentials," small voltage changes in the cell receiving a signal (see also section on downregulation in Chapter Thirteen) (Gillies and Allen, 1977; Pascuzzo, Akaike, et al., 1984; Anderson, Chamberlain, et al., 1986).
Why Is PB Selected Instead of Other Carbamates? PB is preferred over other carbamates, particularly physostigmine, because it has fewer side effects, longer duration of action, and a greater margin of safety (Kolka, Burgoon, et al., 1991a). Physostigmine has been viewed as a candidate for pretreatment, but it has several disadvantages for field use: it has quite a short half-life in vivo, protecting dosing levels produce side effects, and it penetrates the CNS. CNS penetration leads to concerns that use of physostigmine could result in adverse effects on troops' performance.
PB does not cross the blood-brain barrier under ordinary circumstances, because of its positive charge (Kolka, Burgoon, et al., 1991b). Thus CNS side effects from PB are not thought to be an issue with normal use. While PB does not protect AChE in the CNS from nerve agents, it also presumably does not itself induce CNS symptoms and potentially deleterious performance changes (by inhibiting brain AChE. (Protection of the CNS is done postexposure, using atropine, oximes, and diazepam.) PB's ability to confer protection in doses without substantial side effects has made it the preferred pretreatment over physostigmine (Sidell, 1990).
Additive Effects of PB with Soman Have Not Been Clinically Reported. Because PB and nerve agents both inhibit AChE, it might be supposed that the toxicity would be additive--that a smaller amount of a nerve agent would be needed to produce toxicity if PB were given in advance. For reasons not well understood (which may relate to the ability of PB to desensitize the ACh receptor, as noted above), an additive effect on performance and on lethality evidently does not occur, at least for soman. (Studies in primates suggest that in the short term, prior to three hours after administration, AChE inhibition with soman is greater when PB pretreatment has been given--suggesting some additivity of effect; but in the longer term, beyond six hours, AChE inhibition with soman is substantially less if PB has been given (Blick, Murphy, et al., 1987). The dose of soman that proves lethal is not reduced in animals given PB before a soman challenge (instead, the lethal dose is markedly increased--that is, PB is protective). Moreover, pretreatment with PB in humans administered very small amounts of soman led to no change or fewer effects than without pretreatment (Sidell, 1990), consistent with primate studies in which sublethal doses of soman led to similar or slightly less effects if PB pretreatment had been given (Blick, Murphy, et al., 1987).
Similarly, prolonged miosis (constriction of the pupils) produced by the AChE-inhibiting OP DFP was prevented by pretreatment with physostigmine (a carbamate compound related to PB). The time-course of miosis was shortened to that of physostigmine alone, suggesting that DFP did not attach to the receptor site (Leopold and McDonald, 1948) and was protected from doing so by the carbamate (in this case physostigmine rather than pyridostigmine).
It should be noted, however, that if PB is given after nerve agent rather than before, additive effects might be expected (Pope, 1997).
Efficacy of PB Pretreatment for Soman
One primate study evaluated the effect of soman with and without PB pretreatment: without PB, the AChE inhibition induced by soman was still complete (0 percent functional) at the termination of the study; for PB pretreated animals, though blockade was briefly complete at 10 minutes (0 percent functional), by 30 minutes AChE activity had been restored (Dirnhuber, French, et al., 1979). The time-course of blockade was consistent with that produced by PB alone. In another study, PB pretreatment markedly improved the protection conferred by atropine and oxime in primates: the protective ratio was increased with PB pretreatment from 1.6 to more than 40 in rhesus monkeys (Kluwe, Chinn et al., 1987). Lesser benefits against lethality from soman with PB pretreatment have been seen in other, nonprimate species (see Table 3.5).
| Nerve Agent | PR:a No PB |
PR:a With PB |
Species | Reference |
| Soman (GD) | 1.6 | >40 | Rhesus monkey | Kluwe, Chinn, et al., 1987 |
| 1.5 | 6.4 (5.0b) | Guinea pig | Jones, Carter, et al., 1985 | |
| 2.0 | 2.7 (7.1b) | Guinea pig | Lennox, Harris, et al., 1985 | |
| 1.9 | 4.9 | Guinea pig | Capacio, Byers, et al., 1993; Capacio, Koplovitz, et al., 1995 | |
| 1.7 | 6.8 | Guinea pig | Inns and Leadbeater, 1983 | |
| 1.4 | 2.7 | Rabbit | Joiner, Dill, et al., 1989 | |
| 2.2 | 3.1 | Rabbit | Sultan and Lennox, 1983 | |
| 1.9 | 2.8 | Rabbit | Koplovitz and Stewart, 1994 | |
| 1.1 | 2.5 | Mouse | Sultan and Lennox, 1983 | |
| 1.2 | 1.4 | Rat | Anderson, Harris, et al., 1992 | |
| Sarin (GB) | 36.4 | 35 (24b) | Guinea pig | Koplovitz, Harris, et al., 1992 |
| 2.1 | 2.2 (2.0b) | Mouse | Koplovitz, Harris, et al., 1992 | |
| Cyclosarin (GF) | NA | >5 | Rhesus monkey | Koplovitz, Gresham, et al., 1992 |
| 2.7 | 3.4 | Guinea pig | Stewart and Koplovitz, 1993 | |
| 1.4 | 1.4 | Mouse | Stewart and Koplovitz, 1993 | |
| Tabun (GA) | 4.4 | 7.8 (12b) | Guinea pig | Koplovitz, Harris, et al., 1992 |
| 2.4 | 3.9 | Rabbit | Joiner, Dill, et al., 1989 | |
| 4.2 | >8.5 | Rabbit | Koplovitz and Stewart, 1994 | |
| 1.3 | 1.7 (2.1b) | Mouse | Koplovitz, Harris, et al., 1992 | |
| VX | 58.8 | 47 (45b) | Guinea pig | Koplovitz, Harris, et al., 1992 |
| 7.8 | 6.0 (3.9b) | Mouse | Koplovitz, Harris, et al., 1992 | |
| 2.5 | 2.1 | Rat | Anderson, Harris, et al., 1992 |
SOURCE:
Adapted from Dunn, et al., 1997; and Pyridostigmine, 1996.
aWith atropine plus oxime treatment.
bPR with two doses of PB given.
No studies to assess protection by PB against death from soman, or any other nerve agent, have been done in humans, because experiments involving lethal doses of nerve agent obviously cannot be done in humans.
Efficacy of PB Pretreatment for Other Nerve Agents
As Table 3.5 shows, in rodents PB may slightly improve the protective ratio for tabun and maybe cyclosarin (based on studies in guinea pigs) and may slightly reduce the protective ratio for sarin and VX. Nonetheless, the protective ratios remain well over 5 for these agents, in rodents, so that PB has been perceived as not exerting a particularly concerning detriment on the very good protection provided by atropine and 2-PAM for these agents.
The presumption is that reduction in benefit occurs because both PB and nerve agent are binding to, and inhibiting, AChE. (1) PB confers added immediate detriment for peripheral AChE sites, by binding and inhibiting additional molecules of AChE, over and above those inhibited by nerve agents, with little or no offsetting benefit. Because there is time for oxime to be given after nerve agent exposure and before significant aging can take place (e.g., for sarin-AChE complexes), irreversible AChE inhibition is not expected; the oxime will have pulled off the sarin before the complex has aged. (PB is not needed to block and protect some fraction of the AChE from permanent inactivation, since the inactivation would presumably not be permanent.) (2) Additional peripheral AChE blockade will occur because PB in addition to nerve agent will bind and inhibit AChE, and increased binding of the peripheral sites by PB may "drive" increased central binding by nerve agent. (More nerve agent may be available to cross into the brain, because it has not been tied up by binding to peripheral sites that are already occupied by PB.) Tabun also has a long half-time of aging but may be somewhat less oxime-sensitive, at least in guinea pigs. Whether other mechanisms of protection are in play remains a matter of debate.
The actual evidence regarding the effect of PB on protection against nerve agent lethality is limited, in major part because it is based on studies in animals. Indeed, for most nerve agents, the evidence is based on studies in rodents and lagomorphs, which are not closely related to humans. One study in primates compares protective ratios with and without PB, in the context of postexposure treatment with atropine and pralidoxime (analogous to the regimen available to military personnel); this represents a relatively limited body of data. No studies were identified in which the protective ratio against sarin of standard nerve agent pretreatments were examined with and without PB treatment in primates.
There are differences in results from one study to another within an animal species and marked differences across animal species (differences that are not always in the same direction from one nerve agent to another). PB pretreatment, as previously stated, is presumed to markedly enhance protection against lethality in the event of soman exposure, based on (limited) evidence in monkeys.
PB pretreatment hampers protection against nerve agent lethality for other, nonsoman nerve agents (in the context of atropine and pralidoxime treatment). It is presumed to do so only to a minor and unimportant extent, but this supposition appears to be based solely on evidence from nonprimate animals (Anderson, Harris, et al., 1992; Koplovitz, Harris, et al., 1992). The effects of PB pretreatment on treatment efficacy--enhancement or reduction of treatment benefit--have not been studied for most nonsoman nerve agents in primates, and the PRs that result with PB pretreatment in primates for these other agents are not known. That is, no studies have been identified that assess protection with and without PB pretreatment in primates, in the context of atropine and pralidoxime postexposure treatment, for the nerve agents sarin or tabun; for cyclosarin, the apparent benefit of PB in rodents was reversed to detriment in primates. This offers the problematic possibility, which evidence should be obtained to preclude, that just as there is an exaggerated response to PB for soman pretreatment in primates versus rodents in the direction of increased protection, so there may be an exaggerated response to PB for sarin pretreatment in primates versus rodents in the direction of reduced protection. However, no evidence supports severely reduced protection in primates. Nonetheless, in a war situation, in which both soman and other nerve agents, such as sarin, may represent threats, the possible reduction in sarin protection (of a severity that remains to be quantitated in primates), considered together with the likelihood of sarin (or other nerve agent attack), should be pitted against the possible increase in soman protection multiplied by the likelihood of a soman attack.
The issue of whether a PR of 5 should continue to be considered the consistent goal of nerve agent pretreatment and treatment, across all nerve agents, is not addressed here, but should be reevaluated.
Guinea pigs are presumed to be a good model for primates with regard to nerve agent treatment studies (Wetherell, 1992). For instance, PB provides higher PR for primates and guinea pigs than for other rodents (order: primates, guinea pigs, rabbits, rats (Sidell, 1990; Dirnhuber, French, et al., 1979)), which may relate to the fact that pralidoxime does not confer significant protection against soman in these two species (Inns and Leadbeater, 1983); efforts to standardize testing of nerve agent countermeasures advise using guinea pigs for initial studies followed by nonhuman primates (Koplovitz, Gresham, et al., 1992). Although widely used as models for nerve agent defense studies, guinea pigs represent a problematic model; oxime efficacy is dramatically different in guinea pigs compared to primates for instance, pralidoxime confers benefit against GF in primates but not in guinea pigs, so that the slight benefit PB confers vis-^-vis tabun in guinea pigs might not be present in primates, which appear to be more oxime-sensitive.
Another limitation in current evidence is that the effect of PB in rhesus monkeys is not known to reflect the effect of PB on other primates, and particularly on humans; more confidence would be achieved if studies were performed--and showed similar findings--in other primate species and in species more closely related to humans. There is some theoretical cause for optimism, since aging rates in several species of primates--in marmosets, squirrel monkeys, and cynomolgus monkeys--differ from those in rodents--rats and guinea pigs--in a consistent direction (Talbot, Anderson, et al., 1988), providing some rationale to think that similar effects may occur across primate species. However, there are also several reasons for concern in extrapolation of results from primates. First, doses of protective agents in primate studies have been higher than those in humans; doses of PB three times (Koplovitz, Gresham, et al., 1992), 10 times (Dirnhuber, French, et al., 1977), or even 20 to 50 times higher (Dirnhuber, Green, et al., 1977) than those used in humans have been employed (perhaps most commonly 10 times higher), with the argument that since these higher doses produce AChE inhibition comparable to that with the 30 mg thrice a day regimen in humans, these doses are therefore "physiologically similar" (Dirnhuber, Green, et al., 1978). Atropine doses are also four times higher, due to the "reduced sensitivity" of the monkeys to atropine, which is presumed to also hold for central effects (Dirnhuber, Green, et al., 1978). Results from these different dose studies, which also typically combine the three possible postexposure doses, are used in representing the expected PRs. However, for many reasons these doses cannot be presumed to be physiologically similar.
First, the very fact that higher doses of PB and atropine are needed in nonhuman primates to gain comparability on one physiological measure suggests the monkeys may not represent a good model. Second, it is known that when multiple measures are used to rank treatments, not all measures show effects in the same direction (D'Mello, Cross, et al., 1994); in particular, a higher PB dose may be needed in primates to generate comparable AChE inhibition but may produce highly noncomparable effects in some other measure. This is important because AChE inhibition might not represent the sole mechanism of benefit. Third, in vitro studies in primate and human muscle tissue suggest that 10 times lower, not 10 times higher doses of PB in primates are needed to generate protection against soman in vitro (although this now refers to pipetted doses, rather than oral doses--10-6M is needed in human intercostal muscle tissue to produce somewhat comparable benefit to that of 10-7M PB in primates, although it is possible that this is a function of different sensitivity to, and perhaps incomplete ability to wash off, soman (Smith, 1981). No data were identified that assessed whether comparable oral doses of PB in humans and primates lead to comparable muscle concentrations. If they do, it would imply that the primate studies on which we base estimates of PRs for humans use 100 times the "physiologically equivalent dose" of PB given to humans, if physiological equivalence is defined by the metric of soman protection, presumably the metric of interest. This makes the simplifying assumption of linearity of response. Undoubtedly several of these assumptions, e.g., of comparable muscle concentration after the same weight-adjusted oral dose, will be found to be incorrect. Unfortunately they cannot be assumed to be incorrect in any given direction. This adds substantial uncertainty to estimates of protective benefit of PB even against soman, the nerve agent it is presumed to have protective efficacy against.
In short, the use of PB as a pretreatment adjunct improves survival in tested species of primates with soman exposure, but these tests employ substantially higher mg/kg doses of PB than employed in humans, as well as higher mg/kg doses of atropine; data from other mammals suggest PB does not materially hamper survival with exposure to other nerve agents in these mammals, though there is no direct evidence of this in primates for some nerve agents, including sarin and tabun.
It should be noted that "protection" by PB against lethality from soman attack does not appear to be accompanied by protection against incapacitation from soman, so that mission completion will not necessarily be abetted by PB pretreatment. The aim of maintaining 20 to 40 percent RBC ChE inhibition is "only recommended for counteracting the lethal effects of nerve agent poisoning and are not necessarily appropriate in maintaining combat effectiveness (a desirable characteristic)" (Moylan-Jones, Parks, et al., 1979). Animal studies have reliably shown that even "low" doses of nerve agent, on the order of one to two LD50s, are associated with profound incapacitation despite PB and posttreatment regimens that markedly reduce lethality (Dirnhuber Green, et al., 1978; Moylan-Jones, Parks, et al., 1979; Hayward, Wall, et al., 1990; Wetherell, 1992).
There remain issues regarding whether AChE inhibition by PB (and "protection" of bound sites from nerve agent attachment) actually represents the mechanism of protection by PB. A new drug application for PB as military pretreatment, filed in May 1996, suggests that the degree of AChE inhibition relates to the degree of protection (Pyridostigmine, 1996). Higher doses may produce more average cholinesterase inhibition and more protection, up to a point, and in guinea pigs and rats, a positive correlation exists between the degree of cholinesterase inhibition and protection (Lennox, Harris, et al., 1985). But this need not actually imply a causal connection: at the individual level, the animals with a greater degree of enzyme inhibition do not necessarily experience more protection. In guinea pigs, though PB dose correlated strongly with AChE inhibition, there was no clear relation of the PR to either the PB dose and/or to the level of AChE inhibition (Jones, Carter, et al., 1985). In primates, the level of AChE inhibition was reported not to be critical to effectiveness (Koplovitz, Gresham, et al., 1992). The FDA, in failing to approve the 1996 submission of the New Drug Application, has expressed concern that the evidence from studies in monkeys and guinea pigs does not permit the conclusion that RBC acetylcholinesterase inhibition is either proportional to the degree of protection or necessary for protection:
"the data . . . did not allow us to reach the conclusion that the extent of protection is proportional to the extent of red blood cell acetylcholinesterase inhibition, or even that red blood cell acetylcholinesterase inhibition is a necessary concomitant of protection. To the contrary, there is a dissociation between the two. In the monkey studies, the enzyme inhibition is unrelated to the extent of protection. Furthermore, in guinea pigs, for example in one published study (in Fundamental and Applied Toxicology, 5, S242-S251 (1985)), red blood cell acetylcholinesterase inhibition rates from 5 percent to 80 percent were associated with PRs of 5.0 to 6.4 with little or no suggestion of a dose relationship." (Prendergast, 1997.)
Time-Course of Effectiveness. Studies in guinea pigs show a maximum protective benefit of PB pretreatment when soman exposure occurs 60 minutes after pretreatment. PRs (the ratio of LD50 with treatment divided by LD50 without treatment) for different time intervals following exposure are listed in Table 3.6 (Sidell, 1990; Gordon, Leadbeater, et al., 1978). Because of metabolic and other differences, the time-course in humans may be somewhat different.
| Interval from PB to Soman | Protective Ratio (PR) |
| 10 minutes | 3.4 |
| 30 minutes | 8.0 |
| 60 minutes | 12.5 |
| 2 hours | 6.0 |
| 3 hours | 4.7 |
| 4 hours | 3.2 |
Chemical Warfare Threat. The decision to employ PB as a nerve agent pretreatment was based on the information available regarding the perceived threat of chemical warfare. At least 23 nations are reported to have stockpiles of nerve agents, though these are prohibited for use in war by the 1925 Geneva Convention and the 1989 Paris Convention on Chemical Weapons (which not all nations have signed) (Gunderson, Lehmann, et al., 1992). The nations reported to have chemical weapons include not only Iraq and Iran, but Egypt, Syria, Libya, Israel, Ethiopia, Burma, Thailand, North Korea, South Korea, Cuba, Vietnam, Taiwan, China, and South Africa (Barnaby, 1985; Gunderson, Lehmann, et al., 1992). In addition, the Iraqis not only have CW capability, but have militarized nerve agents and used them against Iranians and against the Kurds (Barnaby, 1985; Gunderson, Lehmann, et al., 1992). Use of nerve agent was confirmed by chemical examination of casualties (Barnaby, 1985). Typically, Iranian casualties were exposed after the explosion of artillery shells or bombs between 4 and 200 meters away (Barnaby, 1985). After 1984, chemical warfare intensified and became more frequent, despite numerous UN missions to investigate use of CW in the Iran-Iraq conflict, and repeated appeals by the UN). Iraq used chemical weapons more extensively and more frequently than Iran. Therefore, CW was justifiably understood to represent a valid Iraqi threat in the PGW. This threat was subsequently confirmed by the discovery of nerve agent munitions, including those containing sarin and cyclosarin, in the Khamisiyah ammunitions storage facility in Iraq (Gulflink, 1997).
Although no direct intelligence indicated that soman stockpiles were present in Iraq, it was known that the former Soviet Union had stockpiled soman. With the dissolution of the Soviet Union and the consequent political and economic destabilization and fragmentation, the military was concerned that arms of many sorts were for sale to the highest bidder, possibly including Iraq.
How Nerve Agents Work. Nerve agents include the G agents (GA or tabun; GB or sarin; GD or soman); and VX. The G agents are viewed as "nonpersistent" since they evaporate and disperse over several hours in temperate conditions. The oilier VX is construed as "persistent" since it may linger for weeks or longer, continuing to pose a hazard (Dunn, 1989). Other nerve agents such as cyclosarin (GF), or thiosarin, are derivatives of these G nerve agents. Nerve agents work by "irreversibly" inhibiting AChE (that is, with very long half-life), as described above, leading to excess cholinergic activity in the brain and the "periphery" (skeletal muscles, smooth muscles and glands), producing respiratory failure and death.
How Long Have Carbamates Been Used for AChE Inhibitor Pretreatment? The first use of a carbamate for pretreatment for lethal effects of an organophosphorus compound was in 1946; physostigmine was reported to protect cats against an otherwise lethal dose of the organophosphate DFP (Koster, 1946). The first use of a carbamate as pretreatment for a nerve agent occurred later, in sarin-challenged rats. Atropine alone was unsuccessful as a therapy (one of five rabbits lived), but pretreatment with physostigmine led to enhanced survival (five of five rabbits lived) (Wills, 1963; Sidell, 1990). PB was considered a candidate pretreatment drug as early as the mid-1950s (Wannarka, 1984), and a major effort to develop a useful formulation was under way at the Biomedical Laboratory at Edgewood, Maryland, in the early 1970s (Wannarka, 1984), though enthusiasm for chemical warfare defense waned in the mid 1970s, stalling progress. An IND (investigational new drug) application for PB for pretreatment use was filed with the FDA in February of 1984 (Wannarka, 1984). Subsequent interactions between the FDA and DoD on this issue are detailed in a separate RAND report (Rettig, 1999).
Side Effects
In two decades of PB testing in the military, the incidence of side effects with PB administration was reputedly under 1 percent. Most side effects were mild, principally involving the GI tract (increased flatus, loose stool).
In pre-PGW military tests of PB, there is some suggestion that incidence of side effects may not have been as low as supposed; use of small samples may have obscured even relatively pervasive effects. For example, in one study containing a list of 38 symptom questions, each at four times, in a small set of subjects, it was stated that no symptoms were significantly more common in those assigned to PB compared to a placebo (Gleadle, Kemp, and Wetherell, 1983b). In fact, however, symptom reporting was typically more common in PB patients compared to placebo patients. Symptoms were reported more often in PB than placebo cases 97 times, compared to 18 times with placebo patients reporting a symptom at a higher rate than PB patients. The ratio becomes 100:18 if one includes the query to patients that they attributed the symptoms to their pill; the results, using the sign test, are z/2 = 7.37, p < .0001. If each time period is not considered separately, the resulting value is z/2 = 4.96, p < .001. This suggests that across a wide range of symptoms, in patients blinded to PB versus placebo status, symptom reporting was indeed significantly more common in those receiving PB (Gleadle, Kemp, and Wetherell, 1983b).
The PGW experience entailed a substantially higher incidence of reported side effects than those anticipated from pre-PGW studies. As part of the legal requirement for use of PB as an investigational new drug, DoD was required to collect data on the safety and efficacy of PB (Federal Register, 1997). Three surveys have been conducted to determine the incidence and severity of side effects associated with the use of PB as a nerve agent pretreatment. The first was a questionnaire sent to 42 selected medical personnel involved in Operation Desert Shield and Operation Desert Storm; 23 of the questionnaires were completed and returned. Among the 23, 10 responded that the drug was tolerated either very well or well. The most common side effects reported were gastrointestinal (abdominal cramps, nausea, and diarrhea), with other effects including weakness, light-headedness, exacerbation of asthmatic symptoms, fatigue, sleep disturbances, and reduced mental concentration. Of the many thousands of military personnel reported on,[2] eight were hospitalized for side effects attributed to PB, with reasons for hospitalization including cholelithiasis (gallstones), asthma, and allergic skin reaction (Federal Register, 1997).
The second survey was a questionnaire given to an unspecified number of soldiers deployed in the PGW; 149 responded. Of those who took the drug, 37.5 percent reported side effects, most frequently gastrointestinal. Nausea was reported most frequently (11 percent of subjects), followed by headache (7.5 percent) (Federal Register, 1997).
The third survey was designed to document the effect of PB on aviators' ability to carry out combat missions. Of the 118 aviators who participated, 48 were taking other medications, most commonly the antibiotic ciprofloxacin, and 26 of the 108 who reported taking the drug experienced side effects they attributed to PB, most commonly headaches and diarrhea (Federal Register, 1997).
In one published retrospective study, 30 medical officers from the Army's XVIII Airborne Corps were questioned regarding symptoms in personnel they oversaw during the PGW. These officers oversaw 41,650 soldiers and 234,000 person-days of PB use and were reportedly in daily close contact with these units (Keeler, Hurst, et al., 1991). Based on their recall, 50 percent of troops had GI changes (including flatus, loose stools, abdominal cramps, and nausea). Urinary urgency or frequency was also common. Less than 5 percent complained of headaches, runny nose, sweating, and tingling of the extremities. However, these symptoms did not noticeably interfere with performance of "the full range of demanding physical and mental tasks required" (Keeler, Hurst, et al., 1991).
Few (one percent) perceived a need for medical attention and about 0.1 percent discontinued PB based on medical advice. A total of 483 aid station or clinical visits related to PB occurred, usually within hours of taking the first tablet. In some patients, these effects continued as long as PB was taken. In others, it abated after one to two days of use. Reported problems leading to medical attention are listed in Table 3.7.
| Complaint | Number |
| GI disturbances severe enough to prompt medical visits | 313 |
| Urinary frequency | 150 |
| Bad dreams | 5 |
| Worsening of acute bronchitis | 3 |
| Headache | 3 |
| Slurred speech but normal neurological exam | 3 |
| Rashes, one of these with urticaria (hives) of hands and feet | 2 |
| Vertigo | 1 |
SOURCE: Keeler, Hurst, et al., 1991.
Later information from hospital personnel indicated that two women of low weight (45-50 kg) reported increased salivation, severe abdominal cramping, nausea, sweating, and muscle twitching. The presence of symptoms in these individuals was taken to suggest a possible effect of body weight, since lower-weight individuals receiving the same fixed dose in fact are subject to a higher dose-per-body-weight. During the PGW, administration of PB with meals was reported by some to reduce GI complaints and was instituted by some units (Keeler, Hurst, et al., 1991).
A second published report included both a cross sectional study of 213 Israeli soldiers from one unit, regarding symptoms and severity 24 hours after starting PB; and a case-control comparison of AChE inhibition, with the same survey questionnaire, for nine soldiers with and 12 without complaints from a different unit (Sharabi, Danon, et al., 1991; see Table 3.8). The most frequent symptoms were nonspecific and included dry mouth, malaise, fatigue, and weakness. The more predictable symptoms were infrequent, including nausea, abdominal pain, frequent urination and runny nose. Symptoms were mild, and there was no correlation of subjective symptoms with BChE inhibition (Sharabi, 1991).
Another study (unpublished), by the British Medical Services of 208 British PGW personnel, found that 25 percent of the study group suffered side effects serious enough to consider discontinuation of PB (Martin, 1994). More than half missed at least one tablet in first 48 hours of treatment--"a time when one would have expected compliance to be close to 100%." It is noted that "Anecdotal evidence suggests that symptoms attributable to NAPS [nerve agent pretreatment system] therapy may continue for some time after cessation of treatment." These studies of individuals who took PB during the PGW are observational and uncontrolled but may reflect the PGW experience.
Studies outside the PGW venue are more likely to be controlled but may lack the potential interactions between PB and other exposures, including stress. (In addition, individual reports of adverse responses that could reflect individual differences have been discounted in some studies. In one study, it was stated that "A man [in the PB group] who complained of persistent fatigue, headache, poor concentration and irritability had domestic problems" (Kemp and Wetherell, 1982), as though causality can be imputed. Finally, the nature of the "placebo" chosen was stated in only one study identified; the placebo was lactose (Moylan-Jones, Parkes, et al., 1981). Lactose is known to cause gastrointestinal problems among members of the population who are "lactose intolerant" (and this has led to problems in placebo-controlled trials in the past in which lactose was employed as a placebo) (Golomb, 1995); a true increase in GI and perhaps other symptoms could be obscured by employment of lactose as a placebo.
One double-blind crossover study of seven soldier volunteers who received one week each of a placebo and of PB, with daily exposure to four hours of heat, two hours of rest, and two hours of moderate exercise (40 percent maximum aerobic power), failed to detect differences in symptoms (based on a questionnaire) between PB and placebo, though the PB group demonstrated reduced hand grip strength (p <.05), higher rectal temperature (p <.01) and smaller pupil diameter (p < .01) (Cook, Kolka, et al., 1992).
Another study followed 45 male and 45 female subjects in various weight categories who received 30 mg of PB or a placebo every eight hours, for 22 days (Lasseter and Garg, 1996). Controls reported more headaches than the active treatment group (10 of 30, p < .05). While diarrhea and abdominal pain were reported by four of 15 subjects in each active treatment group, the difference between PB and placebo in this small sample was not significant (p = .068).
Less directly germane is information on nine PGW veterans who attempted to overdose on PB, consuming 13 to 30 tablets of 30 mg of PB (390-900 mg) (Almog, Wingler, et al., 1991). Findings included the following:
Findings relating to physiological parameters at rest or with exercise include no change (Sidell, 1990) or a reduction (Kolka and Stephenson, 1990; Stephenson and Kolka, 1990; Gall, 1981; Sidell, 1990; Lasseter and Garg, 1996) in heart rate (of about five beats per minute); no change (Gall, 1981; Sidell, 1990) or reduction (Cook, Kolka, et al., 1992) in pupil size; no change (Roberts, Sawka, et al., 1994) or increase (Kolka and Stephenson, 1990; Stephenson and Kolka, 1990; Cook, Kolka, et al., 1992) in temperature; no change (Glikson, Achiron, et al., 1991) or reduction (Cook, Kolka, et al., 1992) in motor strength; no change (Sidell, 1990) or increase (Kolka and Stephenson, 1990; Stephenson and Kolka, 1990) in sweating; and no change or reduction (Kolka and Stephenson, 1990; Stephenson and Kolka, 1990) in skin blood flow. Effects may vary by ambient temperature, work load, duration of exercise, and other factors.
Acute effects are indirectly relevant to the issue of possible delayed and chronic effects, but are not directly the focus of this report. Nonetheless, Appendix B shows results of several studies of the effects of PB in humans on physiological parameters, performance, and side effects.
[2]The source cited states that 5,825 personnel were reported on. Dr. Ronald Clawson, in reviewing this report, states, "There is a misconception that the number of personnel reported on in this study was 5,825. Due to unclear wording in the survey, the responders identified the number of people who worked for them (i.e., medical personnel) rather than the number of people in the units that they supported. The survey forms were mailed to the commanders of every major medical unit in the theater, covering hundreds of thousands of personnel."